
近日,天津大学团队制备出阳离子空位工程化硅酸锰(V-Mn₂SiO₄)催化剂,其降解罗丹明 B 和 2,4 - 二氯苯酚的速率常数分别达 1.171 min⁻¹ 和 0.1291 min⁻¹,可比于原子分散金属纳米催化剂。该团队通过实验与 DFT 计算证实,V-Mn₂SiO₄中 vacancy 邻近 Mn 位点激活过一硫酸盐,O 位点吸附污染物,使罗丹明 B 180 秒降解率达 96.2%,2,4 - 二氯苯酚 12 分钟降解率达 100%。
该成果以 “Enhancing Reactive Oxygen Species Generation and Pollutant Adsorption in Advanced Oxidation Processes with Cation Vacancy-Driven Dual-Site Mn Catalysts” 为题发表在《Advanced Materials》期刊
内容简介
本研究聚焦高级氧化工艺(AOPs)中活性氧(ROS)生成量低、利用效率不足的问题,提出阳离子空位驱动的双位点 Mn 催化剂设计方案。通过热诱导相变过程中钴的脱溶,成功合成具有阳离子空位的硅酸锰(V-Mn₂SiO₄)催化剂,该催化剂具备 Mn 和 O 双反应位点。实验与密度泛函理论(DFT)计算表明,空位附近的 Mn 位点是过一硫酸盐(PMS)激活的主要活性中心,可高效生成单线态氧(¹O₂),邻近 O 位点则能促进污染物吸附。在催化性能上,V-Mn₂SiO₄对罗丹明 B(RhB)的降解速率常数为 1.171 min⁻¹,是纯 Mn₂SiO₄的 10.3 倍,对 2,4 - 二氯苯酚(2,4-DCP)12 分钟内降解率达 100%,且经过 4 次循环后仍保持良好稳定性。同时,降解 intermediates 毒性显著降低,RhB 和 2,4-DCP 的矿化效率分别达 83.50% 和 64.52%,为环境修复领域提供高效、可持续的催化材料解决方案。
研究背景
高级氧化工艺(AOPs)凭借高活性氧(ROS)降解有机污染物,成为应对环境污染的重要策略,其中多相过渡金属基催化 AOPs 因操作可行性和适应性受关注。然而,其实际应用受限于催化剂和氧化剂前体用量大,核心问题是现有催化剂本征催化活性低,导致环境修复中资源消耗过多。
催化 AOPs 作为级联催化体系,包含催化剂激活前体(如 H₂O₂、过二硫酸盐、过一硫酸盐 PMS)生成 ROS,以及污染物通过直接氧化或 ROS 二次反应降解两个关键过程,因此性能优化需提升催化位点生成 ROS 的本征活性和 ROS 利用效率。
提升本征催化性能可通过调控过渡金属 d 带中心、优化吸附能、促进电子转移实现,但 ROS 在水介质中寿命极短(如・OH<1μs、SO₄⁻<30-40μs),需高效时空耦合才能有效氧化污染物。此外,空位工程尤其是阳离子空位设计,能直接调控活性位点几何和电子结构,在过渡金属基催化剂中引入阳离子空位被认为是提升 AOPs 性能的有效策略,且深入理解阳离子空位反应特性对指导新型催化剂开发至关重要。基于此,本研究设计阳离子空位锚定的硅酸锰催化剂,以实现 AOPs 中 ¹O₂生成与污染物吸附的协同增强。
研究内容
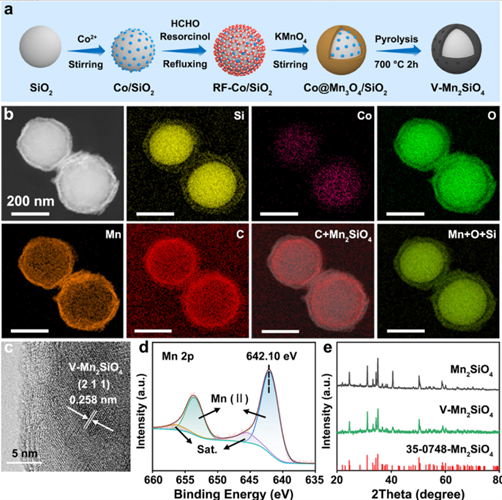
图 1 V-Mn₂SiO₄催化剂的制备过程、元素分布、微观结构、XPS 及 XRD 表征
该图系统展示了 V-Mn₂SiO₄催化剂的制备与关键表征结果。图 1a 为核壳结构 V-Mn₂SiO₄催化剂的制备流程,以 SiO₂为起始原料,经 Co²⁺改性得到 Co/SiO₂,再包覆间苯二酚 - 甲醛(RF)形成 RF-Co/SiO₂,加入 KMnO₄后形成 Co@Mn₃O₄/SiO₂,最终在 700℃下热解 2 小时,通过钴脱溶生成 V-Mn₂SiO₄。图 1b 的元素 mappings 显示,V-Mn₂SiO₄中 Si、Co、O、Mn、C 元素分布清晰,核心为 SiO₂,Mn₂SiO₄壳层中 Mn、Si、O 均匀分布,C 和 Co 位于核壳界面,证实核壳结构形成。图 1c 的高分辨透射电镜(HRTEM)图像显示,V-Mn₂SiO₄存在 0.258 nm 的晶格间距,对应 Mn₂SiO₄的(211)晶面,表明其良好的晶体结构。图 1d 的 Mn 2p X 射线光电子能谱(XPS)显示,结合能在 653.56 eV(Mn 2p 1/2)和 641.67 eV(Mn 2p 3/2)处有特征峰,峰间距 11.6 eV,拟合结果证实 Mn(II)为主要氧化态。图 1e 的 X 射线衍射(XRD)图谱中,V-Mn₂SiO₄在 24.7°、31.2°、35.0° 和 50.7° 处出现四个主要特征峰,分别对应 Mn₂SiO₄的(021)、(031)、(211)和(042)晶面(JCPDS No.35-0748),与纯 Mn₂SiO₄对比,进一步验证 V-Mn₂SiO₄的成功合成及晶体结构完整性。
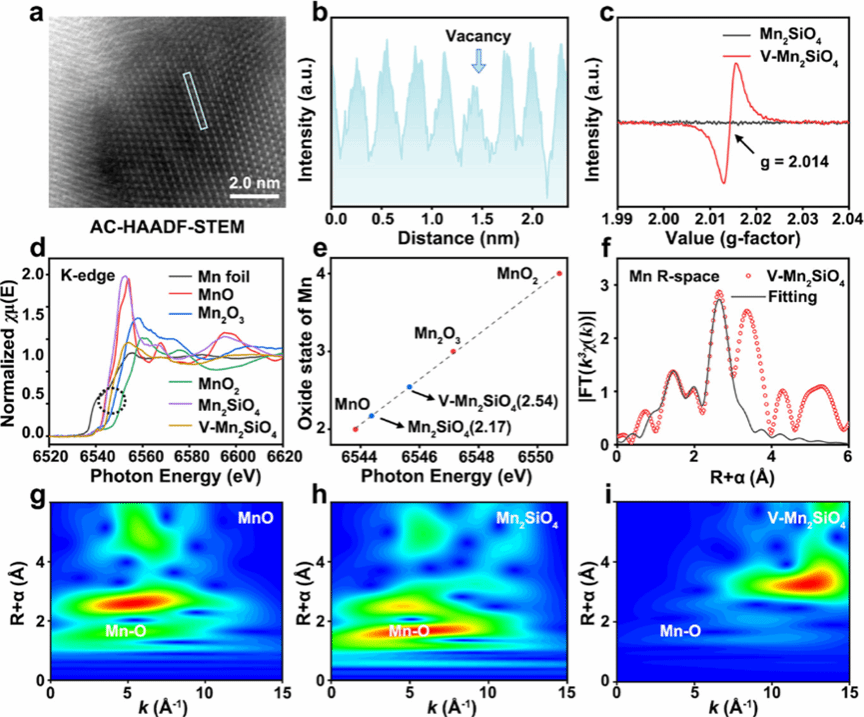
图 2 V-Mn₂SiO₄中阳离子空位的表征
该图从多个维度证实 V-Mn₂SiO₄中阳离子空位的存在及相关电子结构变化。图 2a 的像差校正高角环形暗场扫描透射电镜(AC-HAADF-STEM)图像显示 V-Mn₂SiO₄的晶格条纹,可观察到晶格中存在原子缺失位点,初步指示阳离子空位。图 2b 对应的 Mn 原子强度分布图中,特定位置出现强度骤降,进一步证实阳离子空位的存在。图 2c 的电子顺磁共振(EPR)谱图显示,V-Mn₂SiO₄在 g=2.014 处有强信号,对应阳离子空位产生的未成对电子,而纯 Mn₂SiO₄无明显信号,明确证明 V-Mn₂SiO₄中存在阳离子空位。图 2d 的 X 射线吸收近边结构(XANES)光谱表明,V-Mn₂SiO₄和 Mn₂SiO₄的 Mn K 边介于 MnO 和 Mn₂O₃之间,说明 Mn 氧化态在 + 2 至 + 3 之间,且 V-Mn₂SiO₄的 K 边向高能量方向偏移。图 2e 计算得出 V-Mn₂SiO₄中 Mn 的价态为 2.54,高于 Mn₂SiO₄的 2.17,与 XPS 结果一致,表明阳离子空位导致电荷密度重新分布。图 2f 的 Mn K 边扩展 X 射线吸收精细结构(EXAFS)拟合曲线显示,V-Mn₂SiO₄的 Mn-O 配位数为 4.2,低于 Mn₂SiO₄的 4.6。图 2g-i 的小波变换(WT)EXAFS contour 图进一步证实 V-Mn₂SiO₄中 Mn 物种的配位数更低,从结构层面确认阳离子空位的存在及较高的空位浓度。
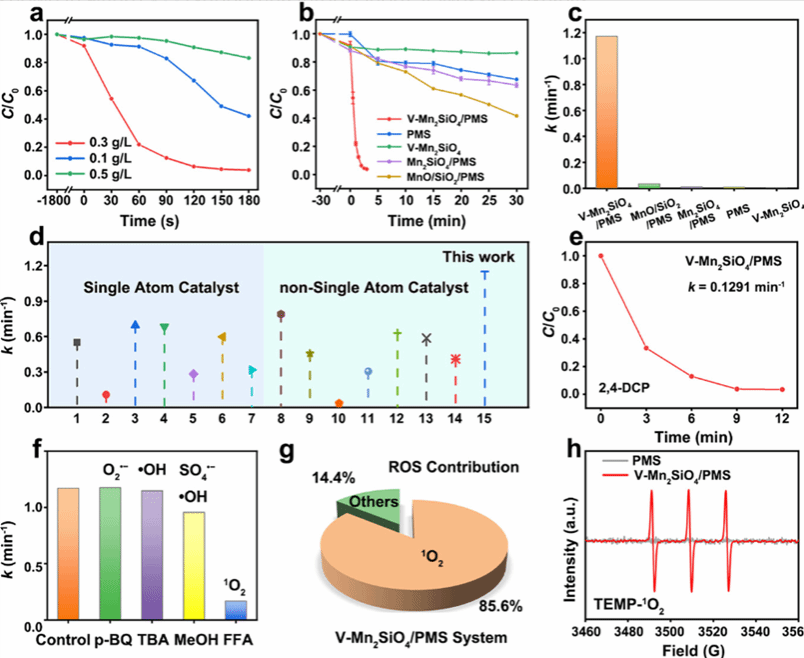
图 3 V-Mn₂SiO₄催化剂的催化性能评价
该图全面评估了 V-Mn₂SiO₄催化剂在污染物降解中的催化性能及相关机制。图 3a 考察 Co 前驱体浓度对 RhB 降解的影响,当 Co 前驱体浓度为 0.3 g/L 时,V-Mn₂SiO₄在 180 秒内对 RhB 的降解率达 96.2%,远高于 0.1 g/L(16.8%)和 0.5 g/L(57.9%)浓度下的降解率,对应速率常数 1.171 min⁻¹,分别是 0.1 g/L 和 0.5 g/L 浓度下的 21.7 倍和 4.0 倍,确定 0.3 g/L 为最佳 Co 前驱体浓度。图 3b 对比不同反应体系对 RhB 的降解效果,仅 V-Mn₂SiO₄与 PMS 共存时,RhB 降解效率显著,180 秒内达 96.2%,而 MnO/SiO₂/PMS 体系 30 分钟降解率 58.4%,Mn₂SiO₄/PMS 体系 30 分钟降解率 36.5%,单独 PMS 或 V-Mn₂SiO₄降解效果微弱,证明 V-Mn₂SiO₄与 PMS 的协同催化作用。图 3c 的速率常数对比显示,V-Mn₂SiO₄/PMS 体系的速率常数(1.171 min⁻¹)是 MnO/SiO₂/PMS(0.0346 min⁻¹)的 33.8 倍,是 Mn₂SiO₄/PMS(0.1138 min⁻¹)的 10.3 倍。图 3d 将 V-Mn₂SiO₄与其他已报道的单原子和非单原子催化剂对比,其速率常数处于较高水平,凸显优异的 PMS 激活能力。图 3e 显示 V-Mn₂SiO₄/PMS 体系对 2,4-DCP 的降解曲线,12 分钟内降解率达 100%,速率常数为 0.1291 min⁻¹,证明对难降解有机污染物的高效降解能力。图 3f 的猝灭实验表明,加入单线态氧(¹O₂)猝灭剂糠醇(FFA)后,RhB 降解速率常数从 1.171 min⁻¹ 降至 0.1688 min⁻¹,而・OH、SO₄⁻和 O₂⁻的猝灭剂对降解影响微弱。图 3g 计算得出 ¹O₂对降解反应的贡献达 85.6%,确定 ¹O₂为主要活性物种。图 3h 的 EPR 谱图显示,V-Mn₂SiO₄与 PMS 共存时,TEMP 捕获剂产生强的三重峰信号,证实 ¹O₂的生成,进一步验证主要活性物种为 ¹O₂。
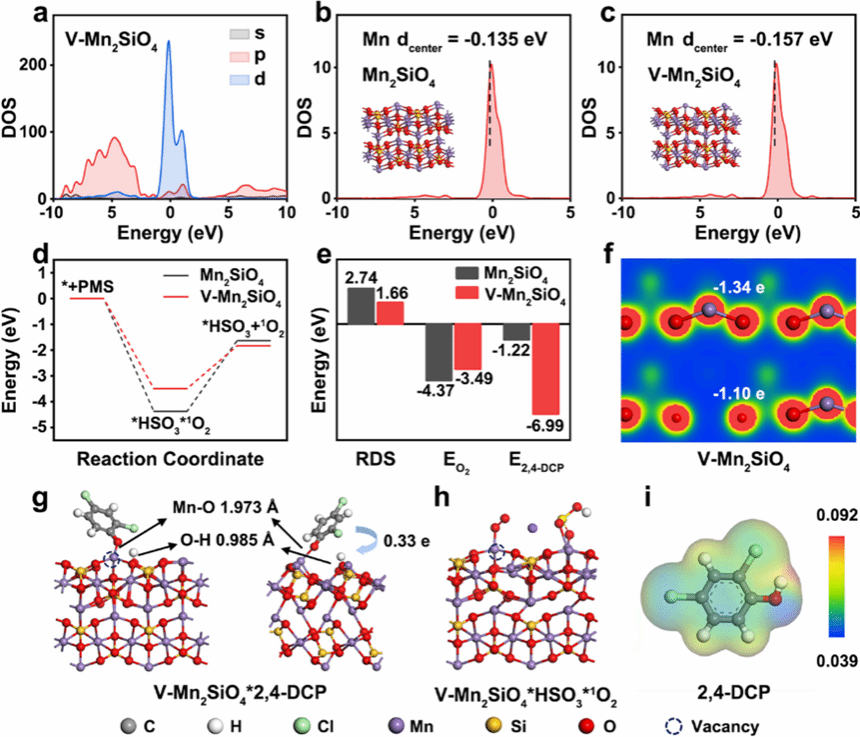
图 4 V-Mn₂SiO₄催化 PMS 激活的 DFT 计算分析
该图通过密度泛函理论(DFT)计算揭示 V-Mn₂SiO₄催化 PMS 激活生成 ¹O₂的机制及双位点作用。图 4a 为 V-Mn₂SiO₄中 Mn 的总态密度(DOS)图,反映电子在不同能量区间的分布情况。图 4b 和 4c 分别为 Mn₂SiO₄和 V-Mn₂SiO₄中 Mn 的投影态密度(PDOS),插入图为对应的计算模型,结果显示 V-Mn₂SiO₄中 Mn 的 d 带中心为 - 0.157 eV,较 Mn₂SiO₄的 - 0.135 eV 降低 0.022 eV,表明 V-Mn₂SiO₄对吸附质的吸附能力较弱。图 4d 的自由能曲线展示 PMS 激活生成 ¹O₂的反应路径,包括 PMS 吸附、解离生成 ¹O₂和 ¹O₂脱附三个步骤,两种催化剂中脱附步骤均为速率决定步骤(RDS),但 V-Mn₂SiO₄体系各步骤的自由能变化更利于反应进行。图 4e 对比 Mn₂SiO₄和 V-Mn₂SiO₄在速率决定步骤(¹O₂脱附)的自由能,V-Mn₂SiO₄的脱附自由能(1.66 eV)显著低于 Mn₂SiO₄(2.74 eV),且 PMS 吸附和解离能也更低,从热力学角度证明 V-Mn₂SiO₄更利于 ¹O₂生成。图 4f 为 V-Mn₂SiO₄的表面电子密度等值面图,显示阳离子空位诱导周围氧原子的电子分布变化,Mulliken 电荷分析表明 V-Mn₂SiO₄中氧原子电荷(-1.10 e)高于 Mn₂SiO₄(-1.34 e),氧原子作为布朗斯特碱位点可捕获质子。图 4g 为 V-Mn₂SiO₄吸附 2,4-DCP 的计算模型,Mn-O 键长 1.937 Å,短于 Mn₂SiO₄中的 2.296 Å,2,4-DCP 在 V-Mn₂SiO₄上的吸附能(-6.99 eV)远高于 Mn₂SiO₄(-1.22 eV),证明 V-Mn₂SiO₄对污染物更强的吸附能力,且存在 0.33 e 从 2,4-DCP 向 V-Mn₂SiO₄的电荷转移,高于 Mn₂SiO₄体系的 0.07 e,表明更强的污染物 - 催化剂相互作用。图 4h 为 V-Mn₂SiO₄同时吸附 HSO₅⁻和 ¹O₂的模型,显示 ¹O₂在吸附 2,4-DCP 附近的 Mn 位点生成,这种空间构型提升 ¹O₂利用效率。图 4i 的静电势(ESP)分析表明 2,4-DCP 具有易被 V-Mn₂SiO₄中富电子 O 位点捕获的官能团,从电子作用角度解释污染物吸附机制,证实双位点协同提升催化效率。
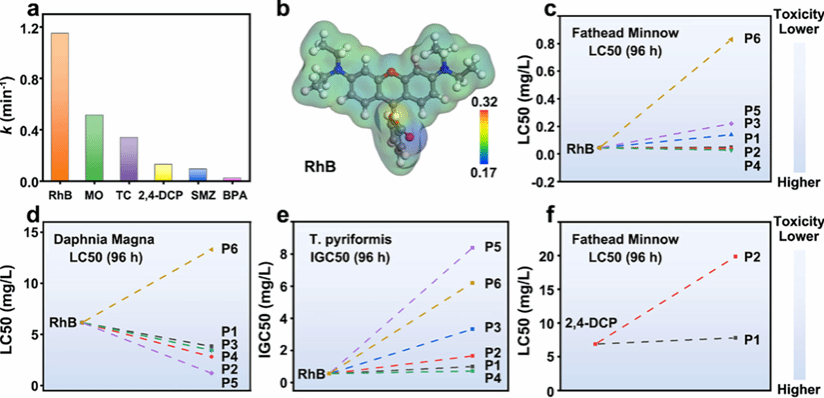
图 5 V-Mn₂SiO₄催化剂的实用性及降解 intermediates 毒性评估
该图验证 V-Mn₂SiO₄催化剂的实际应用潜力及降解过程的环境安全性。图 5a 考察 V-Mn₂SiO₄/PMS 体系对多种模型污染物的降解效果,3 分钟内对甲基橙(MO)、双酚 A(BPA)、磺胺甲噁唑(SMZ)、四环素(TC)的降解率分别达 79%、5.5%、27.2%、66.8%,对应的速率常数分别为 0.5130、0.0216、0.0938、0.3380 min⁻¹,且对 RhB/MO 混合体系 3 分钟内实现完全降解,MO 优先降解,证明催化剂对不同污染物的广谱降解能力及实际混合污染体系处理潜力。图 5b 的 ESP 分析显示 RhB 具有富电子官能团 / 区域,利于与 V-Mn₂SiO₄中 Mn 空位周围的正静电势区域产生静电相互作用,促进局部吸附和电子转移,解释其最高降解速率的原因。图 5c-e 通过 QSAR 模型预测 RhB 降解 intermediates 的毒性,fathead minnow(96 h)LC50、大型蚤(48 h)LC50 和梨形四膜虫(48 h)IGC50 值表明,多数 intermediates 具有较高 LC50/IGC50 值,毒性低于 RhB。图 5f 预测 2,4-DCP 降解 intermediates 的 fathead minnow(96 h)LC50 值,其中 P1 和 P2 的 LC50 值较高,毒性较低;进一步分析显示,RhB 及其 intermediates 虽为发育毒性物质,但 V-Mn₂SiO₄/PMS 体系生成的 intermediates 毒性多低于 RhB;2,4-DCP 及其 intermediates 为 “发育无毒” 物质,且致突变性降低,整体毒性下降,结合 RhB(83.50%)和 2,4-DCP(64.52%)较高的矿化效率,证实该催化体系在环境修复中兼具高效性和安全性。
结论与展望
本研究通过热诱导相变法成功合成富含阳离子空位的硅酸锰(V-Mn₂SiO₄)催化剂,阳离子空位的引入显著改变了 Mn₂SiO₄的几何和电子结构,使锰 / 氧位点在高级氧化工艺(AOPs)中协同发挥双活性中心作用。其中,Mn 位点负责激活过一硫酸盐(PMS)生成活性氧(尤其是单线态氧 ¹O₂),而阳离子空位邻近的 O 位点因局部电子富集,可有效增强污染物吸附。这种空间耦合设计缩短了活性氧与目标污染物的扩散距离,大幅提升活性氧利用效率。
催化性能测试表明,V-Mn₂SiO₄对罗丹明 B(RhB)的降解速率常数达 1.171 min⁻¹,是纯 Mn₂SiO₄(0.1138 min⁻¹)的 10.3 倍,180 秒内 RhB 降解率达 96.2%;对 2,4 - 二氯苯酚(2,4-DCP)12 分钟内降解率达 100%,速率常数为 0.1291 min⁻¹,且催化剂经 4 次循环后仍保持良好稳定性和结构完整性,金属溶出量远低于地表水环境质量标准。此外,降解 intermediates 毒性显著降低,RhB 和 2,4-DCP 的矿化效率分别达 83.50% 和 64.52%,证实该催化剂的环境安全性和高效性。
本研究深入揭示了阳离子空位及界面协同作用在 AOPs 中的作用机制,为设计高效、可持续的环境修复催化材料提供了重要理论指导和实践参考。未来可进一步拓展该空位工程策略至其他过渡金属基催化剂体系,优化催化剂制备工艺以提升空位浓度和稳定性,同时探索其在实际复杂废水处理中的应用,推动高级氧化技术的工业化进程。
参考文献
https://doi.org/10.1002/adma.202513231




